先天性内耳畸形亦称先天性迷路畸形,是胚胎发育早期(胚胎第3-23周)基因突变、缺失或其它遗传因素,以及母亲妊娠期间病毒(细菌、螺旋体)感染、药物(氨基糖苷类、反应停)、理化因素(如X射线)等非遗传因素导致的内耳发育停止或变异,是造成先天性聋的重要原因。
先天性内耳畸形可单独发生,亦可伴随外耳、中耳畸形,部分病例伴有颜面器官、眼、口、齿畸形和(或)伴有肢体与内脏畸形,耳部畸形仅为综合征中的部分表征。
根据2017年Sennaroglu等最新内耳畸形分类标准,可将先天性内耳畸形归为以下几个大类。为了便于理解和加深认识,现将最新内耳畸形分类标准配以部分畸形图片分列如下,以飨读者。
一、内耳畸形
1. 完全性迷路发育不全(Michel畸形):耳蜗、前庭、半规管、耳蜗导水管缺如。

2. 原始耳囊:不完全的听囊残迹,无内听道。
3. 耳蜗未发育
3-1 耳蜗未发育伴前庭正常
3-2 耳蜗未发育伴前庭扩张
4. 共同腔畸形:一个圆形或卵圆形囊性结构代表耳蜗和前庭。
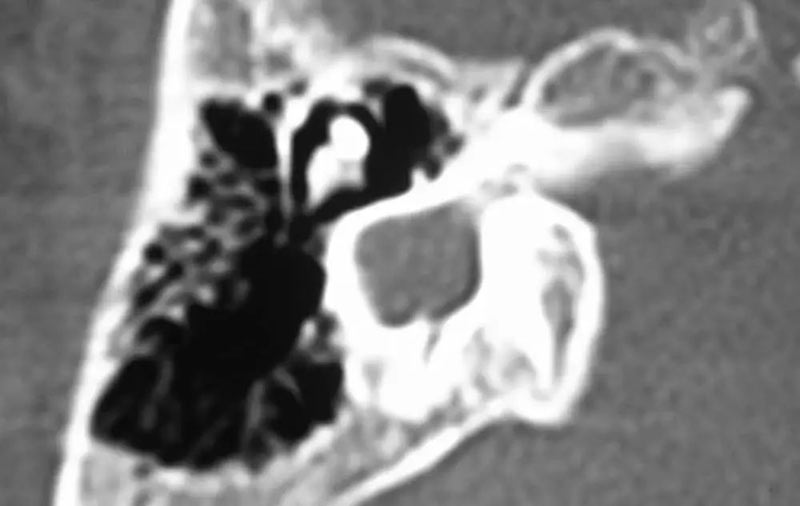
5. 耳蜗发育不全(CH):耳蜗与前庭分隔正常,但比正常耳蜗小。
5-1 CH-1(芽孢样耳蜗)
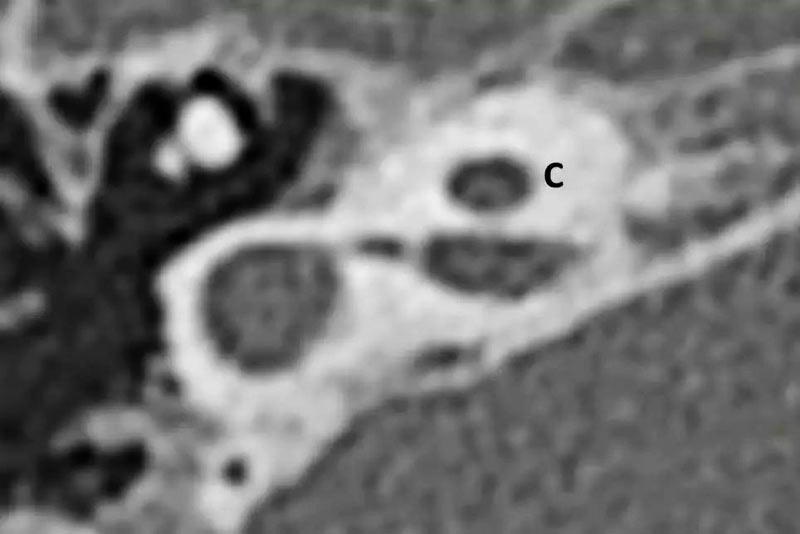
5-2 CH-2(囊性发育不全耳蜗)
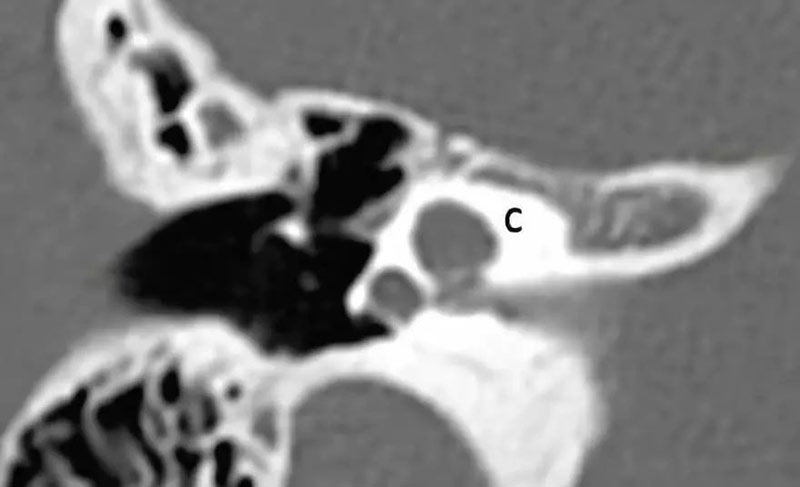
5-3 CH-3(耳蜗少于2回)
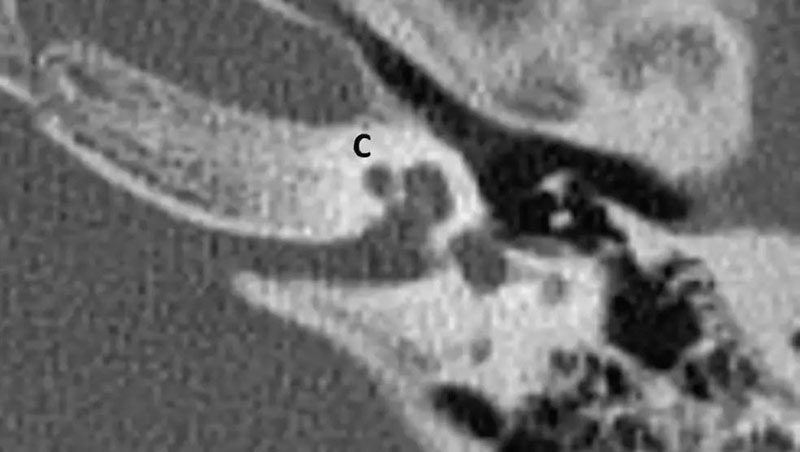
5-4 CH-4(耳蜗中回和顶回发育不全)
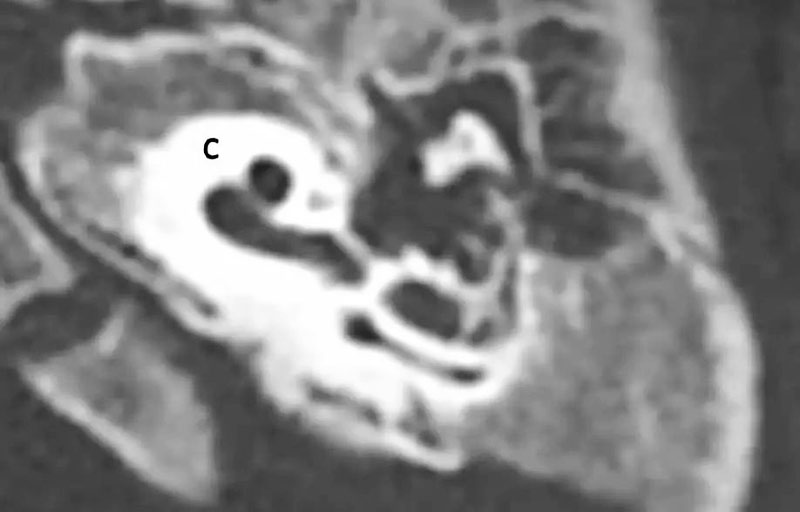
6、耳蜗分隔不全(IP)
6-1 不完全分隔I型(IP-I):囊性耳蜗前庭畸形,无耳蜗蜗轴和筛区(耳蜗与内听道间的区域),导致囊性的外观,伴有一大的囊性前庭。
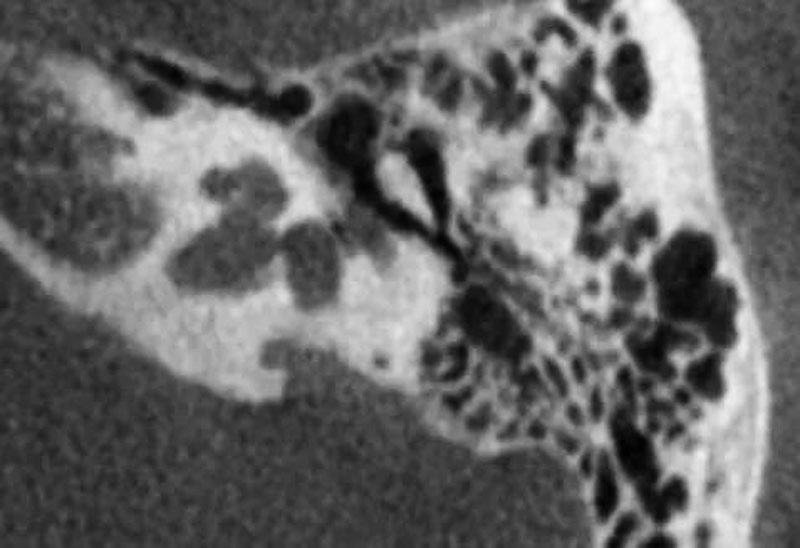
6-2 不完全分隔II型(IP-II):即Mondini畸形,耳蜗包含1.5回,有部分蜗轴,中回和顶回融合构成一囊腔,伴有扩大的前庭和扩大的前庭导水管。
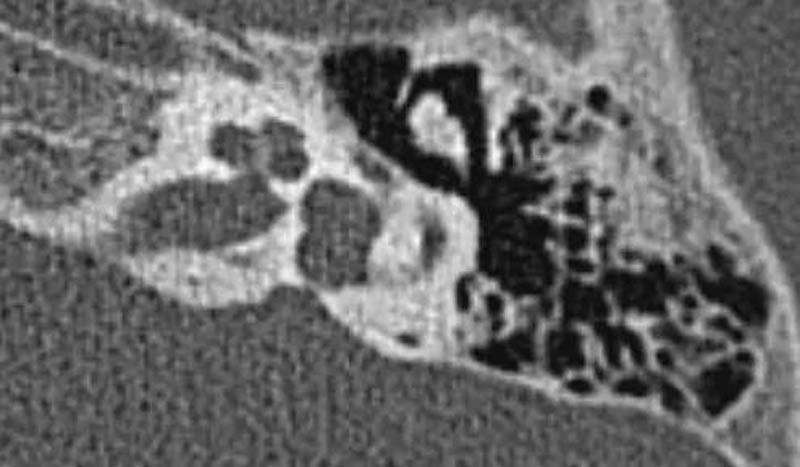
6-3 不完全分隔III型(IP-III):为X连锁遗传性聋。耳蜗大小与正常耳蜗相似,蜗管内间隔正常但全部蜗轴缺乏,耳蜗呈“葫芦形”囊状,内听道与耳蜗相通。
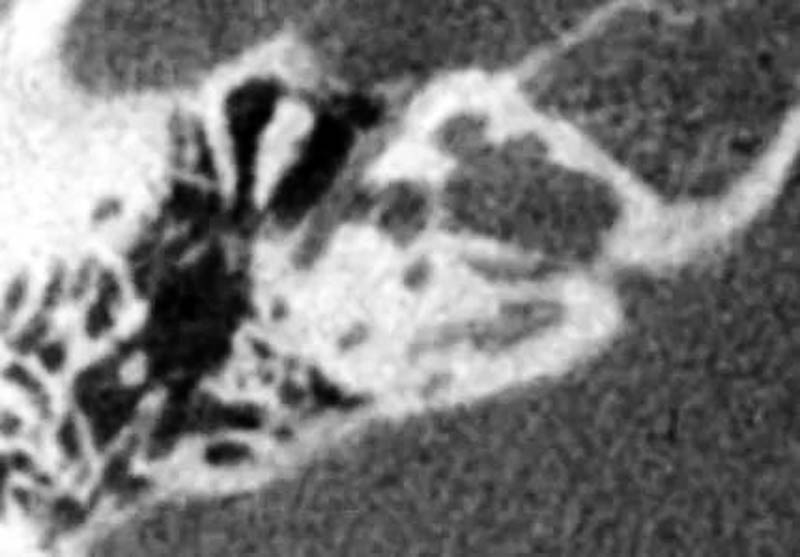
7、前庭导水管扩大
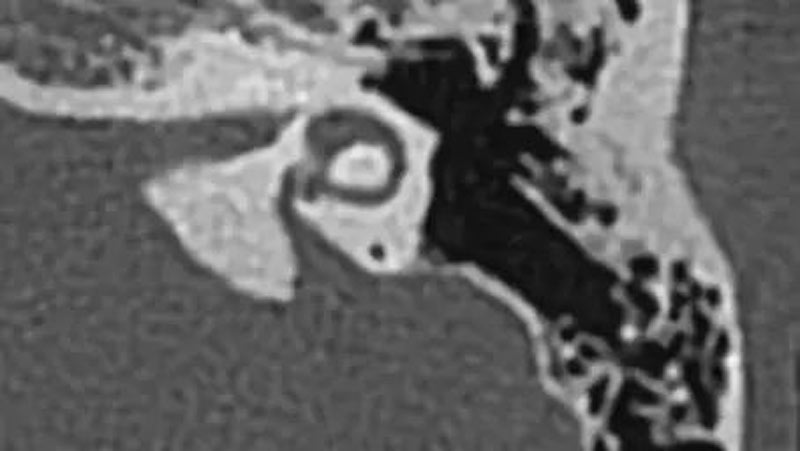
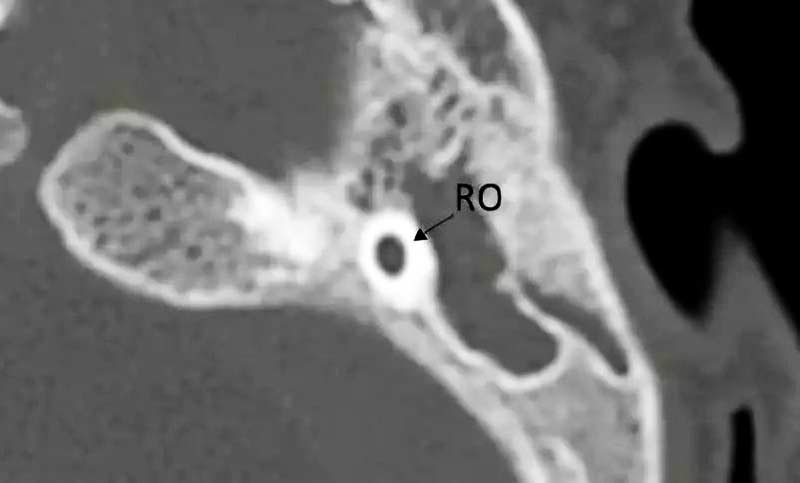
二、骨性耳蜗神经管狭窄或缺失
1. 耳蜗神经管狭窄
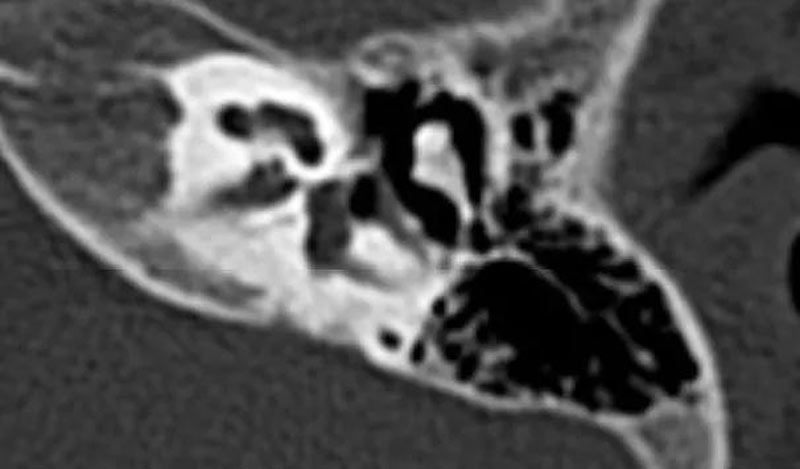
2. 耳蜗神经管缺失

三、耳蜗神经异常
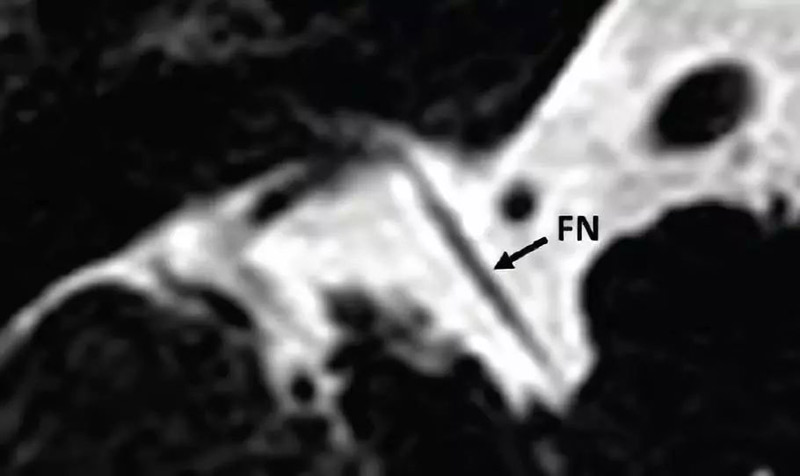
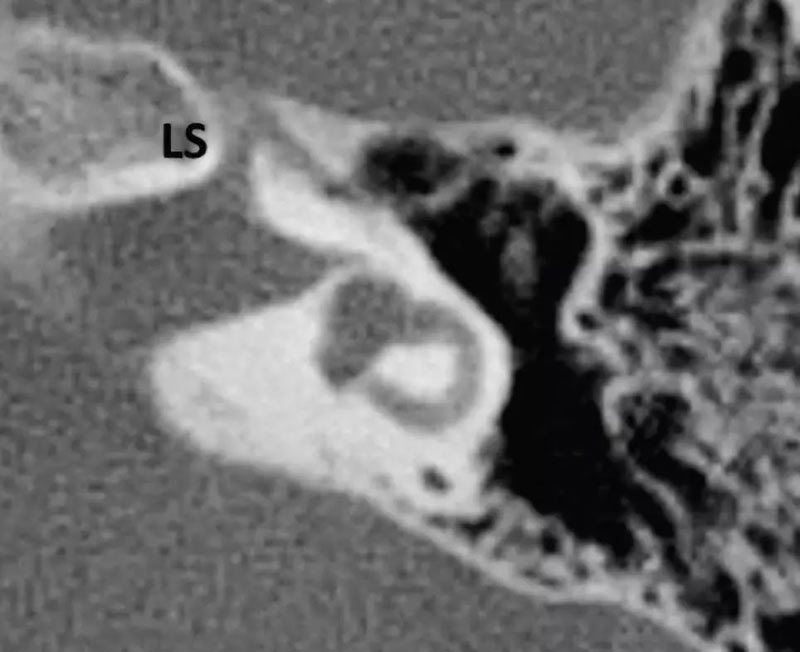
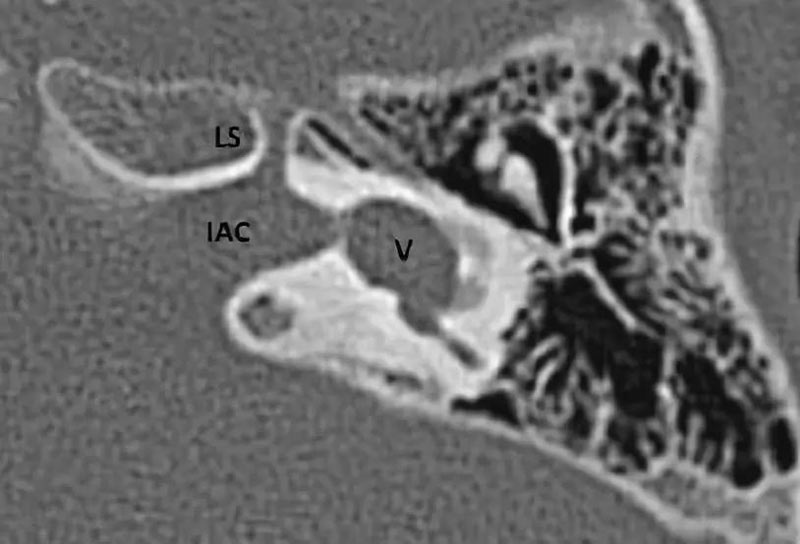
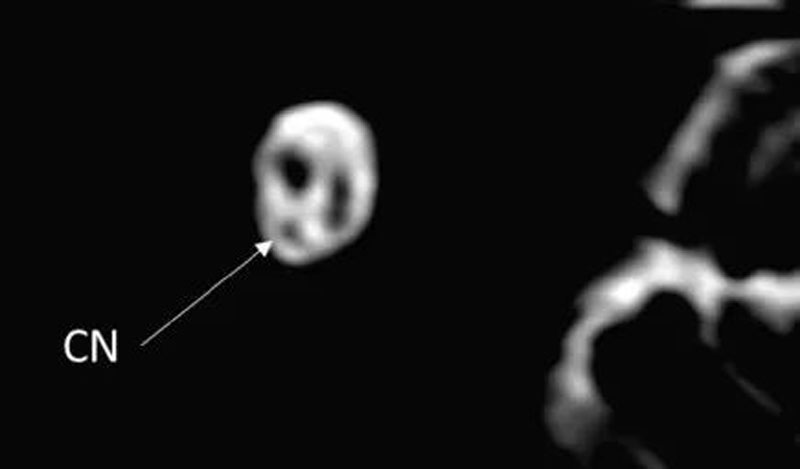
1. 正常耳蜗神经:在旁矢状位,耳蜗神经独立位于内听道前下部分,和同侧面神经大小一致或略大。
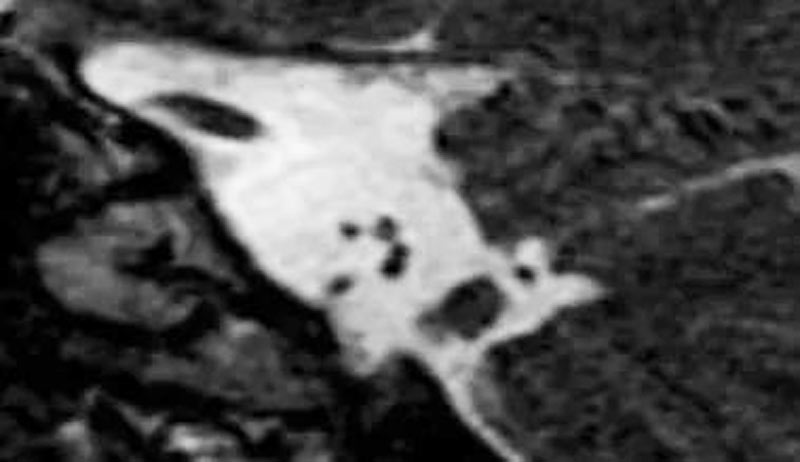
2. 耳蜗神经发育不良:比对侧正常耳蜗神经或同侧面神经小。
3. 耳蜗神经缺如:内听道前下部分无神经,见于耳蜗缺如、耳蜗神经管缺如或发育不良。

4. 正常耳蜗前庭神经:为同侧面神经1.5-2倍或与对侧正常耳蜗前庭神经相似可视为正常。

5. 耳蜗前庭神经发育不良:比对侧耳蜗前庭神经或同侧面神经小。
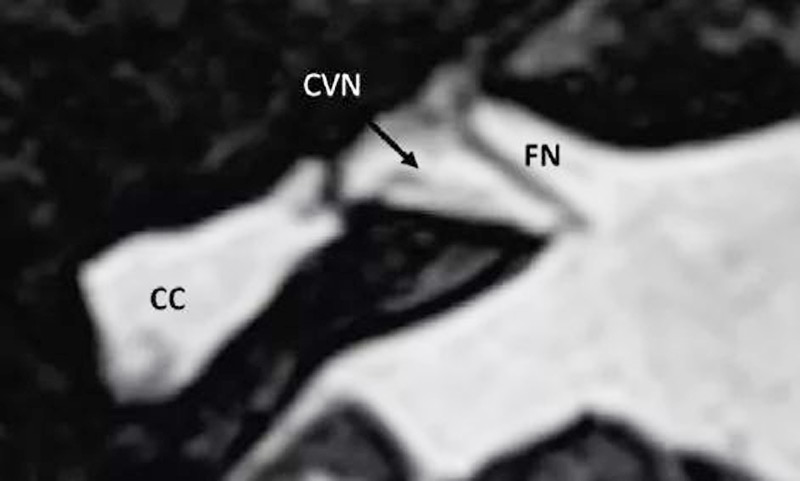
6. 耳蜗前庭神经缺如:Michel畸形内听道缺如,则耳蜗前庭神经缺如,只有面神经存在。